


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
千原 順三*; 山極 満
Progress of Theoretical Physics, 111(3), p.339 - 359, 2004/03
被引用回数:5 パーセンタイル:37.3(Physics, Multidisciplinary)密度汎関数理論は外部ポテンシャルの下での相互作用系の特性を、対応する非相互作用系と関連付けることにより計算する簡便な手法を提供する。ここでは、この非相互作用系の幾つかの関係式を見いだし、中性の電子-原子核混合系に対する熱力学関係式を、非相互作用系の諸量及び交換相関効果を用いて記述する。これにより、原子核に及ぼされる力の定理が容易に証明される。
神林 奨; 千原 順三
Molecular Simulation, 16, p.31 - 46, 1996/00
被引用回数:4 パーセンタイル:17.31(Chemistry, Physical)従来のカーパリネロの理論による分子動力学法とは異なった、新しい第1原理的分子動力学法(QHNC-MD法)を考案した。QHNC-MD法では、液体金属中の電子及びイオンに関する動径分布関数と有効イオン間ポテンシャルに対する量子的HNC方程式を、古典的分子動力学シミュレーションを用いて解く方法である。この方法では、分子動力学シミュレーションによって得られるイオン間分布関数と有効イオン間ポテンシャルを自己無撞着に決定することが可能である。また、QHNC-MD法による有効イオン間ポテンシャルの収束計算は高速であり、しかも、数千から数万個の規模のシミュレーションが可能である。この点はカーパリネロ手法と大きく異なる部分である。液体アルカリ金属に関するQHNC-MDシミュレーションから得られた静的構造因子は、X線・中性子線実験の結果と極めて良く一致し、従来のQHNC方程式の近似解に見られる欠点を取り除くことが可能となった。
千原 順三; 石飛 昌光*
Molecular Simulation, 12(3-6), p.187 - 195, 1994/00
被引用回数:3 パーセンタイル:14.84(Chemistry, Physical)イオン電子混合系とみなせる液体金属においては、その分子動力学を行うとき、常に2体の原子間ポテンシャルで正確に記述できることを示した(多体力は不要)。
大和田 謙; 鈴木 和弥
Spectrochimica Acta, Part A, 50(6), p.1057 - 1063, 1994/00
赤外・ラマンスペクトルデータの基準振動解析から得られる二次のポテンシャル定数(力の定数)を応用して、多原子分子形成時の電子化学ポテンシャルを求める方法を検討した。本報では密度汎関数理論を採用して、これに単純結合一電荷モデル(Simple Bond-Charge Model)を試験的に組み込んだ近似法を提案した。この方法を用いて、種々の異核二原子分子及び多原子分子の電子化学ポテンシャルを計算し、実験値及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。
大和田 謙
Journal of Physical Chemistry, 97(9), p.1832 - 1834, 1993/00
被引用回数:10 パーセンタイル:41.34(Chemistry, Physical)密度汎関数理論において、分子中の各原子の電子エネルギーが電子数と核ポテンシャルについて汎関数展開できることを利用し、多原子分子系の電子化学ポテンシャルを評価するための基本式を導いた。これを用いて種々の多原子分子の電子化学ポテンシャルを計算し、実験値及びサンダーソンの原理から得られる値と比較した結果、基本式の有用性を確かめることができた。
大和田 謙
Spectrochimica Acta, Part A, 49(1), p.81 - 94, 1993/00
異核=原子分子の基準振動解析から得られる調和および非調和ポテンシャル定数を応用して、分子の化学ポテンシャルを求める方法を検討した。本法は主として密度汎関数理論に基礎を置き、分子中の各原子のエネルギーが電子数と核ポテンシャルに関して展開できることを利用する。これらの展開は、N個の電子数と与えられた核ポテンシャルv(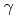 )をもつ系の基底状態の全エネルギーがN,v()の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核=原子分子の化学ポテンシャルを計算し、Sandersonの原理及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。また見掛けの化学ハードニス及び福井関数を含む積分項の値を評価した。分子形成時の化学ポテンシャル変化についても簡単に議論した。
)をもつ系の基底状態の全エネルギーがN,v()の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核=原子分子の化学ポテンシャルを計算し、Sandersonの原理及びab-initio SCF計算から得られる値と比較した結果、本法の有用性を確かめることができた。また見掛けの化学ハードニス及び福井関数を含む積分項の値を評価した。分子形成時の化学ポテンシャル変化についても簡単に議論した。
大和田 謙
Journal of Physical Chemistry, 96(14), p.5825 - 5829, 1992/00
被引用回数:2 パーセンタイル:11.73(Chemistry, Physical)異核二原子分子の基準振動解析から得られる調和および非調和ポテンシャル定数を応用して、分子形成時の各原子の化学ポテンシャル変化と分子の化学ポテンシャルを求める方法を検討した。本法は主として密度汎関数理論に基礎を置き、分子中の各原子のエネルギーが電子数と核ポテンシャルに関して展開できることを利用する。これらの展開は、N個の電子数と与えられた核ポテンシャルv(F)をもつ系の基底状態の全エネルギーがN,v(F)の汎関数として表されるので可能となる。本法を用いて、多数のアルカリハライド分子、その他の異核二原子分子の原子および分子の化学ポテンシャルを計算し、Sandersonの原理から得られる値と比較した結果、本法の有用性を確かめることができた。
